Abstract
Background
Viral genomes contain records of geographic movements and cross-scale transmission dynamics. However, the impact of regional heterogeneity, particularly among rural and urban centers, on viral spread and epidemic trajectory has been less explored due to limited data availability. Intensive and widespread efforts to collect and sequence SARS-CoV-2 viral samples have enabled the development of comparative genomic approaches to reconstruct spatial transmission history and understand viral transmission across different scales.
Methods
We proposed the spatial transmission count statistic that efficiently summarizes the geographic transmission patterns imprinted in viral phylogenies. Guided by a time-scaled tree with ancestral trait states, we identified spatial transmission linkages and categorized them as imports, local transmissions, and exports. These linkages were then summarized to represent the epidemic profile of the focal area.
Results
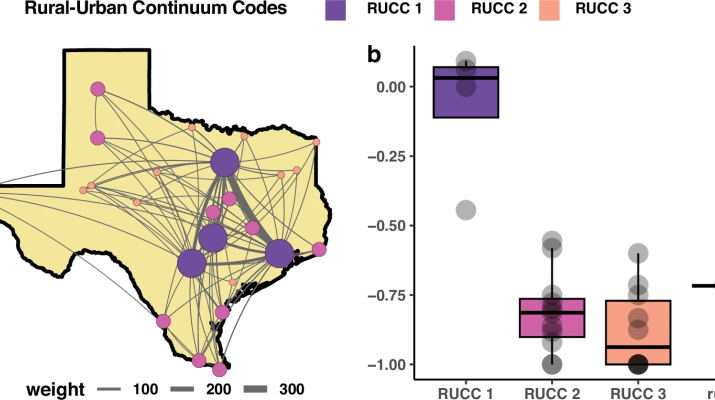
Here, we demonstrate the utility of this approach for near real-time outbreak analysis using over 12,000 full genomes and linked epidemiological data to investigate the spread of SARS-CoV-2 in Texas. Our findings indicate that (1) highly populated urban centers were the main sources of the epidemic in Texas; (2) outbreaks in urban centers were connected to the global epidemic; and (3) outbreaks in urban centers were locally maintained, while epidemics in rural areas were driven by repeated introductions.
Conclusions
In this study, we introduce the Source Sink Score, which determines whether a localized outbreak serves as a source or sink for other regions, and the Local Import Score, which assesses whether the outbreak has transitioned to local transmission rather than being maintained by continued introductions. These epidemiological statistics provide actionable insights for developing public health interventions tailored to the needs of affected areas.